ChIP-seq Pipeline
All Chip-Seq libraries in the database were processed by a unified python scripting pipeline.
To get started, download the script below and input your CSV file with at least two columns: Sample Name, Sample Fastq, Control Name and Control Fastq (if any).
Output includes two folders namely "peakcall" (narrowPeak files) and "BigWig" (bw files).
Overview of the Pipeline
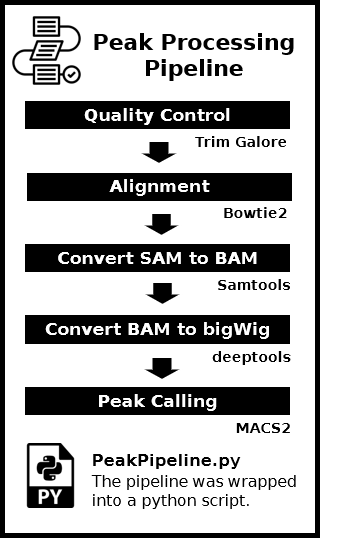
Tool Version of Pipeline
| Tool | Version |
|---|---|
| sratoolkit | 2.11.2 |
| pfastq-dump | 0.1.6 |
| trim-galore | 0.6.4 |
| cutadapt | 2.10 |
| fastqc | 0.11.8 |
| bowtie2 | 2.2.6 |
| samtools | 1.10 |
| macs2 | 2.2.7.1 |
| deeptools | 3.4.3 |
Run the ChIP-seq Pipeline (Docker Version) * Suggested
To execute the ChIP-seq Pipeline Docker version, there is no requirement to configure the environment.
Step 1: Pull the Docker Image
$ unzip TAIR10.zip
1. The file name of the CSV file should be Peak-ProcessTable.csv
Example of Peak-ProcessTable.csv
2. If the sample have multiple SRR files, use ";" to delimite them.
3. If the sample have no input control, left blank.
The file name of the CSV file should be SRAfile.txt
Example of SRAfile.txt
$ docker run -it --rm \
-v /path/of/SRAfiles/:/SRAFiles/ \
dppss90008/qhistone-pipeline \
prefetch --option-file /SRAFiles/SRAfile.txt --output-directory /SRAFiles
-v /path/of/Peak-ProcessTable.csv:/source \
-v /path/of/TAIR10-Bowtie2Index:/Bowtie2Index \
-v /path/of/SRAfiles/:/SRAFiles/ \
dppss90008/qhistone-pipeline \
conda run -n chipseq python /PeakCallingPipeline/Peak-Calling-Pipeline-Version4.py \
--cores 70 \ # CPU cores for speeding up the pipeline \
--bowtie2index TAIR10 \ # bowtie2 index file downloaded from step 2 \
--wkdir /source \
Run Run the ChIP-seq Pipeline (Source Version) Environmental Setting
Please check the following programs are all installed before running the pipeline !!
Step1: Install sratoolkit
$ tar zxvf sratoolkit.current-ubuntu64.tar.gz
$ echo "export PATH=$PATH:/path/to/sratoolkit.3.0.10-ubuntu64/bin" >> ~/.bashrc
$ source ~/.bashrc
$ cd pfastq-dump
$ chmod a+x bin/pfastq-dump
$ wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.12.1.zip
$ unzip fastqc_v0.12.1.zip
$ cd FastQC
$ chmod 755 fastqc
$ sudo ln -s /path/to/FastQC/fastqc /usr/local/bin/fastqc
$ cutadapt --version # Check that cutadapt is installed
$ fastqc -v # Check that FastQC is installed
$ curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/0.6.10.tar.gz -o trim_galore.tar.gz
$ tar xvzf trim_galore.tar.gz
$ tar jxvf samtools-1.19.tar.bz2
$ cd samtools-1.19
$ ./configure
$ make
$ make install
Run the ChIP-seq Pipeline
Step 9: Prepare the ChIP-seq data
2. Store the SRR files in a folder.
1. The file name of the CSV file should be Peak-ProcessTable.csv
Example of Peak-ProcessTable.csv
2. If the sample have multiple SRR files, use ";" to delimite them.
3. If the sample have no input control, left blank.
Change the locations of the programes
INFO = {
"prefetch":"/path/to/sratoolkit.3.0.10-ubuntu64/bin/prefetch",
"pfastq-dump":"/path/to/pfastq-dump/bin/pfastq-dump",
"trim-galore":"/path/to/TrimGalore/trim_galore",
"bowtie2":"/path/to/bowtie2",
"samtools":"/path/to/samtools",
"macs2":"/path/to/macs3",
"bamCoverage":"/path/to/bamCoverage",
"GenomeIndex":"/path/to/Bowtie2Index/genome>
# index file of the genome
"SRA_FILES":"/path/to/ncbi/"
# The folder stored the SRR files
"cores": 70,
# CPU cores for speeding up the pipeline
}